Basic Local Alignment Search Tool (BLAST) Algorithm
(Redirected from BLAST Algorithm)
A Basic Local Alignment Search Tool (BLAST) Algorithm is a sequence alignment algorithm for searching sequence databases.
- Context:
- It can include a family of algorithms such as BLASTP Algorithm and BLASTN Algorithm.
- It can be implemented by a BLAST System that can solve a Sequence Similarity Search Task, particularly Amino Acid Sequences or Nucleotide Sequences.
- …
- Example(s):
- NCBI's Standalone and API BLAST implementation:
NCBI BLAST+(available at: https://blast.ncbi.nlm.nih.gov/Blast.cgi.) - Advanced Biocomputing BLAST (AB-BLAST): https://blast.advbiocomp.com
- QBLAST in BioPython NCBI BLAST module:
Bio.Blast.NCBIWWW.qblast(). - …
- NCBI's Standalone and API BLAST implementation:
- Counter-Example(s):
- See: Approximate String Matching Algorithm, Sequence Homology, Longest Common Subsequence, Shortest Common Supersequence, Longest Common Substring, Shortest Common Superstring, Approximate String Matching, Phylogenetic Analysis Task, Alignment-free Sequence Analysis Task, Levenshtein Distance, Edit Distance, Alignment Distance, Sequential Pattern Mining Task, Dynamic Programming.
References
2021a
- (Kellis et al., 2021) ⇒ Manolis Kellis et al. (2021). "3.5: The BLAST algorithm (Basic Local Alignment Search Tool)". In: LibreTexts (Computational Biology).
- QUOTE: The BLAST algorithm looks at the problem of sequence database search, wherein we have a query, which is a new sequence, and a target, which is a set of many old sequences, and we are interested in knowing which (if any) of the target sequences is the query related to. One of the key ideas of BLAST is that it does not require the individual alignments to be perfect; once an initial match is identified, we can fine-tune the matches later to find a good alignment which meets a threshold score. Also, BLAST exploits a distinct characteristic of database search problems: most target sequences will be completely unrelated to the query sequence, and very few sequences will match.
2021b
- (O'Connor, 2021) ⇒ Clare M. O'Connor (2021). "9.4: BLAST algorithms are used to search databases". In: LibreTexts (Computational Biology).
- QUOTE: BLAST searches begin with a query sequence that will be matched against sequence databases specified by the user. As the algorithms work through the data, they compute the probability that each potential match may have arisen by chance alone, which would not be consistent with an evolutionary relationship. BLAST algorithms begin by breaking down the query sequence into a series of short overlapping “words” and assigning numerical values to the words. Words above a threshold value for statistical significance are then used to search databases. The default word size for BLASTN is 28 nucleotides. Because there are only four possible nucleotides in DNA, a sequence of this length would be expected to occur randomly once in every 428, or 1017, nucleotides, which is far longer than any genome. The default word size for BLASTP is three amino acids. Because proteins contain 20 different amino acids, a tripeptide sequence would be expected to arise randomly once in every 8000 tripeptides, which is longer than any protein. The figure below outlines the basic strategy used by the BLAST algorithms.
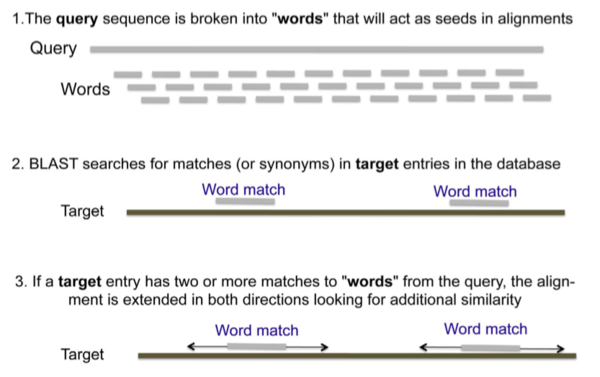
- QUOTE: BLAST searches begin with a query sequence that will be matched against sequence databases specified by the user. As the algorithms work through the data, they compute the probability that each potential match may have arisen by chance alone, which would not be consistent with an evolutionary relationship. BLAST algorithms begin by breaking down the query sequence into a series of short overlapping “words” and assigning numerical values to the words. Words above a threshold value for statistical significance are then used to search databases. The default word size for BLASTN is 28 nucleotides. Because there are only four possible nucleotides in DNA, a sequence of this length would be expected to occur randomly once in every 428, or 1017, nucleotides, which is far longer than any genome. The default word size for BLASTP is three amino acids. Because proteins contain 20 different amino acids, a tripeptide sequence would be expected to arise randomly once in every 8000 tripeptides, which is longer than any protein. The figure below outlines the basic strategy used by the BLAST algorithms.

2021c
- (Wikipedia, 2021) ⇒ https://en.wikipedia.org/wiki/BLAST_(biotechnology) Retrieved:2021-2-25.
- In bioinformatics, BLAST (basic local alignment search tool) is an algorithm and program for comparing primary biological sequence information, such as the amino-acid sequences of proteins or the nucleotides of DNA and/or RNA sequences. A BLAST search enables a researcher to compare a subject protein or nucleotide sequence (called a query) with a library or database of sequences, and identify database sequences that resemble the query sequence above a certain threshold. For example, following the discovery of a previously unknown gene in the mouse, a scientist will typically perform a BLAST search of the human genome to see if humans carry a similar gene; BLAST will identify sequences in the human genomethat resemble the mouse gene based on similarity of sequence.
2020
- (Chang et al., 2020) ⇒ Jeff Chang, Brad Chapman, Iddo Friedberg, Thomas Hamelryck, Michiel de Hoon, Peter Cock, Tiago Antao, Eric Talevich, and Bartek Wilczynski (2020). "Chapter 7 BLAST". In: "Biopython Tutorial and Cookbook".
- QUOTE: We use the function
qblast()in theBio.Blast.NCBIWWWmodule to call the online version of BLAST. This has three non-optional arguments:- The first argument is the blast program to use for the search, as a lower case string. The options and descriptions of the programs are available at https://blast.ncbi.nlm.nih.gov/Blast.cgi. Currently
qblastonly works with blastn, blastp, blastx, tblast and tblastx. - The second argument specifies the databases to search against. Again, the options for this are available on the NCBI Guide to BLAST ftp://ftp.ncbi.nlm.nih.gov/pub/factsheets/HowTo_BLASTGuide.pdf.
- The third argument is a string containing your query sequence. This can either be the sequence itself, the sequence in fasta format, or an identifier like a GI number.
- The first argument is the blast program to use for the search, as a lower case string. The options and descriptions of the programs are available at https://blast.ncbi.nlm.nih.gov/Blast.cgi. Currently
- QUOTE: We use the function
- >>> from Bio. Blast import NCBIWWW
- >>> help(NCBIWWW.qblast)
- ...
- Note that the default settings on the NCBI BLAST website are not quite the same as the defaults on QBLAST. If you get different results, you’ll need to check the parameters (e.g., the expectation value threshold and the gap values).
For example, if you have a nucleotide sequence you want to search against the nucleotide database (nt) using BLASTN, and you know the GI number of your query sequence, you can use:
- Note that the default settings on the NCBI BLAST website are not quite the same as the defaults on QBLAST. If you get different results, you’ll need to check the parameters (e.g., the expectation value threshold and the gap values).
- >>> from Bio. Blast import NCBIWWW
- >>> result_handle = NCBIWWW.qblast("blastn", "nt", "8332116")
2009
- (NCBI, 2009) ⇒ http://blast.ncbi.nlm.nih.gov/Blast.cgi
- The Basic Local Alignment Search Tool (BLAST) finds regions of local similarity between sequences. The program compares nucleotide or protein sequences to sequence databases and calculates the statistical significance of matches. BLAST can be used to infer functional and evolutionary relationships between sequences as well as help identify members of gene families.
2009b
- (Reeves et al., 2009) ⇒ Gabrielle A Reeves, David Talavera, and Janet M Thornton. (2009). “Genome and Proteome Annotation: Organization, Interpretation and Integration.” In: J R Soc Interface, 6(31).
1990
- (Altschul et al., 1990) ⇒ Stephen F. Altschul, Warren Gish, Webb Miller, Eugene W. Myers, and David J. Lipman. (1990). “Basic Local Alignment Search Tool.” In: Journal Molecular Biology, 215.
1981
- (Smith & Waterman, 1981) ⇒ T.F. Smith, and M.S. Waterman (1981). “Identification of Common Molecular Subsequences.” In: Journal of Molecular Biology, 147. (doi:10.1016/0022-2836(81)90087-5).